Thalassemia
Key points
- Thalassemia encompasses a group of hematologic disorders in which alpha- or beta-globin production is reduced or absent. The resulting quantitative red cell disorder is referred to as alpha or beta thalassemia, respectively. This genetic disorder can be caused by one or several gene mutations. Its phenotypes range from silent to fatal.
- All patients with thalassemia should be cared for by a multidisciplinary team who can access red cell transfusion, monitor for cardiac, liver, endocrine and bone complications, and manage such complications as they arise. Access to a hematopoietic stem-cell transplant centre and transition services to adult care should also be considered.
Epidemiology
The maps below show the prevalence of alpha and beta thalassemias.
Figure 1. Global distribution of α- and β- thalassemia
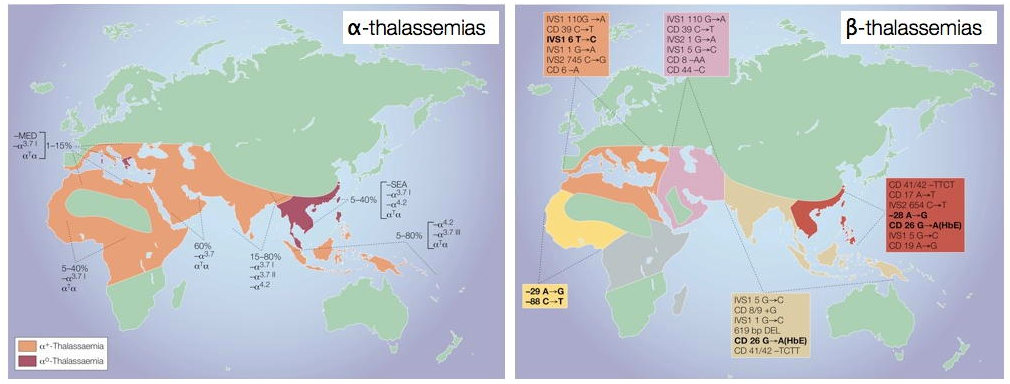
Source: Weatherall, DJ. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nature Reviews Genetics 2001;2(4):245-55.
Alpha thalassemias
Silent carriers of alpha thalassemia — people who have the alpha-thalassemia trait — usually do not require treatment. Their genetic and clinical characteristics are summarized in this table.
|
Variant |
Chromosome 16, |
Signs and symptoms |
|---|---|---|
|
Alpha thalassemia silent carrier |
One of four |
Asymptomatic |
|
Alpha thalassemia trait |
Two of four |
Asymptomatic |
|
Hemoglobin Constant Spring |
Reduced output of alpha globin |
Silent or mildly symptomatic |
|
Alpha thalassemia intermedia with significant hemoglobin H (hemoglobin H disease) |
Three of four |
Moderate-to-severe hemolytic anemia, a modest degree of ineffective erythropoiesis, splenomegaly, variable bone changes |
|
Alpha thalassemia major with significant hemoglobin (Hb Bart’s) |
Four of four |
Causes nonimmune hydrops fetalis, usually fatal |
| Reprinted with permission from Alpha and beta thalassemia, August 15, 2009, Vol. 80, No. 4, American Family Physician Copyright ©2009 American Academy of Family Physicians. All Rights Reserved. | ||
Beta thalassemias
Individuals with the beta-thalassemia trait (one gene affected) usually do not require treatment. Patients who have beta thalassemia major (two beta-globin genes affected) are usually transfusion-dependent from an early age and later require chelation therapy due to subsequent iron overload. Iron supplements should be avoided by people who have thalassemia mutations except when a child has proven iron deficiency. Iron loading is a serious complication of thalassemia major and intermedia.
Preconception genetic counselling should be offered to those at risk of having a child with thalassemia.1
|
Variant |
Chromosome 11, |
Signs and symptoms |
|---|---|---|
|
Beta thalassemia trait |
One |
Asymptomatic |
|
Beta thalassemia intermedia |
Two; mild-to-moderate decrease in beta-globin synthesis |
Variable degrees of severity of symptoms of thalassemia major |
|
Beta thalassemia major |
Two; severe decrease in beta-globin synthesis |
Abdominal swelling, growth retardation, irritability, jaundice, pallor, skeletal abnormalities, splenomegaly; requires lifelong blood transfusions |
| Reprinted with permission from Alpha and beta thalassemia, August 15, 2009, Vol. 80, No. 4, American Family Physician Copyright ©2009 American Academy of Family Physicians. All Rights Reserved. | ||
Management of thalassemia
Canadian guidelines for the care of patients with thalassemia are available.1
All patients with thalassemia should be cared for by a multidisciplinary team who can access red cell transfusion, monitor for cardiac, liver, endocrine and bone complications, and manage such complications as they arise. Access to a hematopoietic stem-cell transplant centre and transition services to adult care should also be considered.
Selected resources
- The Hospital for Sick Children. About Kids’ Health [Handouts]. See What is Thalassemia?
- Richardson M. Microcytic anemia. Pediatr Rev 2007;28(1):5-14.
- Sayani F, Warner M, Wu J, et al, 2009. Guidelines for the clinical care of patients with thalassemia in Canada.
References
- Sayani F, Warner M, Wu J, et al, 2009. Guidelines for the clinical care of patients with thalassemia in Canada.
Reviewer(s)
- Andrea Hunter, MD
- Anna Banerji, MD
Last updated: April, 2018